Some years ago when I was silly enough to have a real job in the chemical industry we were working in a regular synthetic organic lab but had to scale some of our synthetic work up considerably. This makes everything a bit complicated and time consuming. Now you can get a long way by just buying massive RBF's and so forth and as long as you can recrystallise your products everyone is happy. However, when you have to run your 250 grams of stuff down a quick flash column you have a serious problem. So we were looking for some alternative to flash column chromatography that would allow us to do so. A good friend mine at the Australian Wine Research Institute of all places told me about something he called squat columns and gave me a paper from Aldrichimica Acta on the technique:
"Dry-column" Flash Chromatography, L.M. Harwood, Aldrichimica Acta, 1985, 18, p. 25
Now this paper isn't particularly detailed so we had to mess around with it for quite a long time before we got it right but it was worth it. We were columning really large quantities of stuff this way. I spoke to my former industry colleague about it the other day and he told me that they were doing 500 gram columns now collecting 1 and 2 liter fractions in conical flasks. Anyway, we thought that it made sense to publish a paper with more detailed instructions so that someone might actually try to use it successfully. Fortunately, the referees also liked the idea so we got this paper out on the topic: Dry Column Vacuum Chromatography, D.S. Pedersen and C. Rosenbohm, Synthesis, 2001, pp. 2431-2434
Dry Column Vacuum Chromatography, D.S. Pedersen and C. Rosenbohm, Synthesis, 2001, pp. 2431-2434
We even took the liberty of renaming the technique because we thought flash was a bit misleading. Basically Dry Column Vacuum Chromatography (DCVC) is an alternative to flash where the solvent is sucked through the column. The solvent is added one fractions at the time (making very precise gradient elution dead easy) and hence the column is sucked almost completely dry between fractions. For the full details read the paper. So what does it look like? Above there's a picture of a 20 gram column I ran the other day. Now this is a big column but it also works with small columns. My favourite size columns have a diameter of 4-6 cm. They are easy to work with and I would typically collect 20 ml fractions. If you want to give DCVC a go but don't want to ask the glass blower for a fancy piece of glassware at first you don't have to. Some of the bits and pieces you need for a simple set up are shown above. A nice and sturdy spatula is essential for good column packing. Quite a few chemist I know use water aspirators for vacuum rather than a fancy diaphragm pump and it seems to work for them so that's also an option. The trick to make this technique work is to get the right silica.
column but it also works with small columns. My favourite size columns have a diameter of 4-6 cm. They are easy to work with and I would typically collect 20 ml fractions. If you want to give DCVC a go but don't want to ask the glass blower for a fancy piece of glassware at first you don't have to. Some of the bits and pieces you need for a simple set up are shown above. A nice and sturdy spatula is essential for good column packing. Quite a few chemist I know use water aspirators for vacuum rather than a fancy diaphragm pump and it seems to work for them so that's also an option. The trick to make this technique work is to get the right silica.
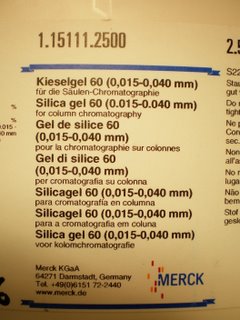 If you use the stuff to the right it will all work fine. However, if you use regular flash silica it will not work. Don't even try it's a waste of time. Regular flash silica is simply to coarse and you can't pack a proper column.
If you use the stuff to the right it will all work fine. However, if you use regular flash silica it will not work. Don't even try it's a waste of time. Regular flash silica is simply to coarse and you can't pack a proper column.
Cool features about DCVC are:
(1) Large scale columns are possible
(2) It's faster than flash. Mainly because you don't collect many fractions (usually 20-30)
(3) You don't use much silica (column is only ~5 cm high) or solvents
(4) Because the column gets sucked dry between fractions you can easily do other stuff whilst running it. Even leaving it for a few hours will not ruin the resolution.
My personal record is seven DCVC's in one day (didn't go home until 10 pm though). However, I wouldn't recommend doing more than four in a day. You start going insane after the fourth column. The technique works so well that I haven't run a flash column in five years and I have no intention of ever doing one again. The columns can normally be recycled. Just give them a good rinse. I usually do something like: MeOH, EtOAc and finally hexane. Seal it up nicely and stick in the fridge. They easily last for a week in the fridge if you seal them up properly. This is how I do it: An old suba seal on the stem a bit of para film on the top with a bit of tape and stick in the fridge in a beaker (see picture).
An old suba seal on the stem a bit of para film on the top with a bit of tape and stick in the fridge in a beaker (see picture).
One important detail that we have discovered since publishing the paper is that one should NOT concentrate the sample to be columned on silica. Instead, you should use Celite (or Kenite). Opposed to silica Celite is very non-polar and hence you can use quite a lot for loading you sample without compromising the resolution. This way you can ensure that you get as much of your compound as possible on to the actual column. If you stick a piece of filter paper on top of the silica column prior to loading the celite it makes it really easy for you to scrape the celite off after running the column so that you can use it once more. Also you should stick a piece of filter paper on top of the kenite so that you don't mess the surface up when you pour the solvent on. Okay I'd better stop here. You obviously need to read the paper before you try this method. Just send an email to curlyarrow@gmail.com to request a copy. D!
"Dry-column" Flash Chromatography, L.M. Harwood, Aldrichimica Acta, 1985, 18, p. 25
Now this paper isn't particularly detailed so we had to mess around with it for quite a long time before we got it right but it was worth it. We were columning really large quantities of stuff this way. I spoke to my former industry colleague about it the other day and he told me that they were doing 500 gram columns now collecting 1 and 2 liter fractions in conical flasks. Anyway, we thought that it made sense to publish a paper with more detailed instructions so that someone might actually try to use it successfully. Fortunately, the referees also liked the idea so we got this paper out on the topic:
 Dry Column Vacuum Chromatography, D.S. Pedersen and C. Rosenbohm, Synthesis, 2001, pp. 2431-2434
Dry Column Vacuum Chromatography, D.S. Pedersen and C. Rosenbohm, Synthesis, 2001, pp. 2431-2434We even took the liberty of renaming the technique because we thought flash was a bit misleading. Basically Dry Column Vacuum Chromatography (DCVC) is an alternative to flash where the solvent is sucked through the column. The solvent is added one fractions at the time (making very precise gradient elution dead easy) and hence the column is sucked almost completely dry between fractions. For the full details read the paper. So what does it look like? Above there's a picture of a 20 gram column I ran the other day. Now this is a big
 column but it also works with small columns. My favourite size columns have a diameter of 4-6 cm. They are easy to work with and I would typically collect 20 ml fractions. If you want to give DCVC a go but don't want to ask the glass blower for a fancy piece of glassware at first you don't have to. Some of the bits and pieces you need for a simple set up are shown above. A nice and sturdy spatula is essential for good column packing. Quite a few chemist I know use water aspirators for vacuum rather than a fancy diaphragm pump and it seems to work for them so that's also an option. The trick to make this technique work is to get the right silica.
column but it also works with small columns. My favourite size columns have a diameter of 4-6 cm. They are easy to work with and I would typically collect 20 ml fractions. If you want to give DCVC a go but don't want to ask the glass blower for a fancy piece of glassware at first you don't have to. Some of the bits and pieces you need for a simple set up are shown above. A nice and sturdy spatula is essential for good column packing. Quite a few chemist I know use water aspirators for vacuum rather than a fancy diaphragm pump and it seems to work for them so that's also an option. The trick to make this technique work is to get the right silica. If you use the stuff to the right it will all work fine. However, if you use regular flash silica it will not work. Don't even try it's a waste of time. Regular flash silica is simply to coarse and you can't pack a proper column.
If you use the stuff to the right it will all work fine. However, if you use regular flash silica it will not work. Don't even try it's a waste of time. Regular flash silica is simply to coarse and you can't pack a proper column.Cool features about DCVC are:
(1) Large scale columns are possible
(2) It's faster than flash. Mainly because you don't collect many fractions (usually 20-30)
(3) You don't use much silica (column is only ~5 cm high) or solvents
(4) Because the column gets sucked dry between fractions you can easily do other stuff whilst running it. Even leaving it for a few hours will not ruin the resolution.
My personal record is seven DCVC's in one day (didn't go home until 10 pm though). However, I wouldn't recommend doing more than four in a day. You start going insane after the fourth column. The technique works so well that I haven't run a flash column in five years and I have no intention of ever doing one again. The columns can normally be recycled. Just give them a good rinse. I usually do something like: MeOH, EtOAc and finally hexane. Seal it up nicely and stick in the fridge. They easily last for a week in the fridge if you seal them up properly. This is how I do it:
 An old suba seal on the stem a bit of para film on the top with a bit of tape and stick in the fridge in a beaker (see picture).
An old suba seal on the stem a bit of para film on the top with a bit of tape and stick in the fridge in a beaker (see picture).One important detail that we have discovered since publishing the paper is that one should NOT concentrate the sample to be columned on silica. Instead, you should use Celite (or Kenite). Opposed to silica Celite is very non-polar and hence you can use quite a lot for loading you sample without compromising the resolution. This way you can ensure that you get as much of your compound as possible on to the actual column. If you stick a piece of filter paper on top of the silica column prior to loading the celite it makes it really easy for you to scrape the celite off after running the column so that you can use it once more. Also you should stick a piece of filter paper on top of the kenite so that you don't mess the surface up when you pour the solvent on. Okay I'd better stop here. You obviously need to read the paper before you try this method. Just send an email to curlyarrow@gmail.com to request a copy. D!


 orcid.org/0000-0003-3926-7047
orcid.org/0000-0003-3926-7047


{kind=link}
39 comments:
Your demonstrator has some beefy forearms. I'd happily read more posts like this - I'll take useful information over gossip/slander/cute stories anyday.
Thanks.
Interesting post. I might try that method next time I've got a lot of product.
I've found this technique works well, but nonpolar stuff tends to shoot right off the column. I'd do this if I had something relatively polar (<0.3 in 30% EtOAc/hexanes) and I was doing it on scale. I see no advantage to doing this on small scale...you have to load your tiny bit of compound on celite, wash all sorts of flasks, and generally go to the trouble of setting it up, whereas you could just dry pack a flash column, separate, and throw away your test tubes in half an hour. Also, the silica you need for this stuff is a lot more expensive, although this should be weighed against the fact that you need a lot less silica, and probably solvent. Some people in our group at Harvard swear by this, and don't ever do flash, but I think that's pretty extreme.
Thanks for the comments guys. Intersting observation about the beefy forearms. I never thougt about that before but I guess it's beefier that your average forearm. Also like everything in life the first time you try it it's probably not going to be perfect so please do a test run on something of less importance before you stick your whatevertoxin B down the column. D!
Very good comment coming from Harvard that raises a couple of good points. (1) DCVC of non-polar compounds: Yes this is indeed a weakness of DCVC. A general rule of thumb about DCVC states that the desired compound will elute in the solvent mixture where it has an Rf value of ~0.5. Hence, when you are dealing with compounds that have high Rf values in even straight hexane the technique is no good if there are any similarly non-polar impurities present. That said I regularly purify very non-polar compounds by DCVC with great success because everything else is in my reaction mixture is much more polar. However, at this point one can argue that I’m no longer running a column but basically just doing a filtration. (2) Small scale columns: To me a column of less than 100 mg of product is small scale and less than 50 mg of product is really small scale. It is not unusual for me to column something in the 50-100 mg range and I've never found it particularly tedious to run DCVC on this kind of scale. However, I do agree that there is no time or cost saving involved between Flash and DCVC when operating on such small quantities. Less than 50 mg of product in my opinion is a Flash job. Nothing beats one of those tiny Pasteur pipette columns when you are messing around with 15 mg of material. (3) Silica cost, Flash silica vs. DCVC silica: If you purchase your silica from Merck a price comparison shows that DCVC silica costs approximately 25% more. Overall when you compare how much solvent and silica you burn using the two techniques DCVC is a lot cheaper. The economical aspect of DCVC is reflected by the people who cite the DCVC paper most. They are generally from financially less endowed institutions/countries and they probably see this as an easy way of saving some cash. (4) And finally: Yes it’s correct that you have to load your compound on celite and this takes a bit of time. For DCVC to be really fast you do things in parallel and several columns in a row. Load your compound on the rotary evaporator and remove the remaining solvent on a high vacuum pump while you pack your column and get everything else ready. Run TLC’s while you do the column. Rinse all the test tubes that don’t contain the desired stuff and recycle them straight away. This way you’ll never need more than 20-30 test tubes for any column. While you are combining fractions and concentrating what your want rinse the column and get the next column going with the next compound that you pre-loaded on celite while you were running the first column. I’ve generally been doing methodology work and hence been in a situation where I could postpone running columns until I had 3-5 compounds needing purification. Only do a maximum of four columns this way or you will become suicidal. D!
Years if gymnastics = beefy forearms
Someone who shall remain anonymous emailed a bunch of questions about DCVC to me that I decided to post here. Also I just had a quick re-read of my paper (not very well written to my disappointment) and realised that there are some more issues I need to address.
(1) Q: Do you think there is much of a problem with cross contamination inside the separatory funnel/apparatus with a stopcock?
A: No….yes…sometimes….it depends on the nature of the stuff you are columning and how you are doing it. Sometimes it will actually crystallise in the receiver. Other times you see an oily residue on the glass. This is usually only ever a problem when you are columning multi grams of material and you can usually see it happening and then deal with it. I basically just take the sintered funnel out and wash the inside of the receiver into the next empty test tube in the rack. The problem is more commonly encountered when using low boiling solvents such as ether or dichloromethane as you are basically concentrating your stuff in the receiver during columning.
(2) Q: Have you found n-heptane to be significantly better in practice, because of higher bp, than hexane or hexanes, as far as evaporation/condensation of moisture?
A: No. n-Hexane or hexanes is fine. The moisture condensation issue was a problem when I was working in Denmark but hasn’t been so while I was in England or here in Australia. So if you work in a high humidity lab it’s a problem otherwise no probs. I use dichloromethane, ether and acetone occasionally without any problems.
(3) Q: Do you always concentrate the sample on Celite, or if it is relatively non-polar, either add it neat (if a liquid) to the silica on top, or dissolve it in hexane, ethyl acetate, or a combination thereof, and then add it to the silica?
A: I tend to always dry load which is a bit silly because it’s not necessary for liquids or non-polar compounds. In other words neat loading of oils/liquids and solvent loading is fine. However, be careful not to use too polar solvents when solution loading as it will mess up your resolution. D!
I was introduced to this technique, Celite modification included, by another Dane who is now at Harvard and is probably the individual mentioned in an earlier post. I love this technique! It's great for large scale! I used it for up to 200 grams in a single go and had no problems! Try that on a flash column! This technique is a lifesaver in total synthesis!
Hi,
let me tell you that I had used this technique with out knowing this was DCVC and was published (may be it was one of the senior grads in the lab had seen the publication). But here is the modification we had. Instead of sep funnel...and testtubes....we always used to have small Erlenmyer flasks (conical falsks as we used to call them in INDIA) with ground joint and couomns with the side arm for vacum filtration (these were nothing but elongated cintered funnels). This technique works great for any scale.
We also used the same silicagel that was purchased for prepartion of TLC plates.
Keep posting and keep up the good work.
shiva
I'm a natural products guy and find myself with 20g of oleoresin. When loading an oil on DCVC are there any volume considerations other than the weight of material? I'm afraid that if I use Celite I'll get a paste that I'll have to spread onto the column... unless that's ok?
PS rf difference is a very generous 0.3 between my compound and the major contaminants.
Much has been written of the relative utility of small and medium scale (<100 g) purifications using this technique.
However, working in Chemical Development (Process Chem) for a large pharmaceutical, I regularly use the technique on 500 g - 1 Kg, applying the same principles already outlined.
I believe it could be used for even larger scale, given the right equipment...but who knows?
Can be much easier than to rid yourself of annoying impurities that cannot be purged through crystallization!!!
Dear Daniel
I though this technique was called VLC? Vacuum Liquid Chromatography. Or at least that what it is called at Copenhagen Uni. But hey, it a great technique and super for big scale things.
Dear sir,
Is it possible to try smaller amounts of stuff (250 mg - 3 gm) by this method? if possible what is the properties of the sintered glass funnel and How much will be the volume of the fractions? Is there any notes about it?
Wow, like someone else said, I had my own go at this technique without any knowledge there were papers published on the matter, or even knowing the technique already existed and had a name.
If I ever have to repeat what I did (six multi-gram columns to get rid of phosphine oxide in one evening) I would definitely consider your points about the Celite and the higher-grade silica. I also agree totally about the suicidal tendencies.
Would it be possible to exchange alumina for the silica gel in DCVC?
That is a very good question. I have very little experience with alumina so I don't know if it's possible. The only alumina I know of is too coarse for DCVC. If you can't make a firmly packed column it just isn't going to work. So to make a long story short it really depends on whether there is small particle size alumnina available. If anyone has tried this please post your comments. D!
Wow.. this is cool.. I have been looking this kind of info for several month.. I'm interested to know how to extract enzyme (hirudin) from leech saliva.. hirudin have similar factor to heparin, anti-coagulant..
D, that is the differnce here between this technique and a simple silica pad. I have recently used a similar technique with a 48cm diameter table top filter and 3Kg of the required grade silica to clean 600g of API for a c-GMP manufacture. We pulled 30 2L fractions and recovered all the material by HPLC assay. Purity post pad is significantly enhanced
Can this technique be used for natural products preparative work?
Another comment from someone stated that:
"EMD is discontinuing product 15111, Silica Gel 60 15-40µm. Does anyone have an alternate supplier for this DCVC stationary phase?
I am testing Sortech's 52200-1 (Silica Gel P - Prep TLC, Premium Rf, 60A, 15-40um, 1kg $ 105.00) to see if it will replace the EMD silica".
Wondering if anyone had figured out if the Sortech product from above works?
@Anonymous, I have worked with Sorbtech's 15-40um Premium Rf silica (among others). It is just as good as Merck's equivalent. Another good company to purchase silica from is Silicycle, check them out.
I tried this using a rather practical setup that does not involve glassblower help, and it seems to work well. Do you have a classic recommendation for tricky compound to separate that can be useful for classroom lab, but shows advantage over flash column ?
I found that celite loading may not be necessary. Set a 1 -1.5 cm thick of sand, pour the compound with minimum solvent around the funnel wall, and load it to the silica layer by pouring solvent works well.
is anybody here? I would like to use this approach with C-18 on silica for stationary phase. I have a large amout of product that I know is contaminated and probably won't crystallize because of this. I can't use plain silica cuz my product is zwitterionic and sticks hard to that surface. I get nice chromatography on reversed phase HPLC so I think I could get rid of thge little junks that come first in this manner. Any suggestions as to particle size? Can I use a slightly taller column (I see one on ChemGlass) instead the short stocky buchner that folks here (and on other sites) seem to use ?
Hey.. I apologize for all my typos and spelling errors in the message above. I must sound like an illiterate high school kid.. sorry for that. I do really want to know if anyone has done this procedure in reversed phase (C-18 on silica). thanks in advance if anyone is still here on this message board.
@Robin: Yes this sort of thing has also been done with RP18 silica. I have done it with the commercially available stuff. The main issue is that it can be hard to pack a tight column because the particles are too big (in my experience). Also some people published a paper 10-20 years ago where they do this. They even describe how to make RP silica by derivatising regular silica.
However, in recent years we have moved on to do this type of large scale RP separations using an automated chromatography system (Combiflash) on commercially available pre-packed RP18 columns. They are really great for doing this type of stuff on gram scale. I hope that helps. D!
thank-you so much for the advice. I have seen the combiflash system and the prepacked redi-sep columns. But I have no $ and need to take the poor person's approach. There is a website for a company called Chemrus
http://www.chemrus.com/. I am wondering if you might comment on their apparatus. I wonder if there is a way to buy the redi-packed columns and use them in some similar type of manual approach.
@Robin: You need to have some kind of pump system if you are going to use the pre-packed C18 columns. The only alternative I can see is to try and pack a column yourself as described in our DCVC paper in Synthesis. In our experience there is no advantage in making columns more than 5-6 cm tall. What matters is the diameter. Higher diameter = more material can be loaded. As I mentioned earlier I have done this myself. I was using a 1 cm diameter column that I packed with RP silica, followed by gradient elution with acetonitrile, water mixtures. D!
D. Thank-you!!! last week when I came back onto the site (a day later)I didn't see my recent message. But it was received!! I am so happy to be getting this info as I am not a trained synthetic chemist but I do these little 1 or 2 pot affairs to make probe molecules and of course the biggest problem is the clean-up. I will endeavor to follow your DCVC approach and hand-pack w/ RP silica. Robin
@Robin I am curious on how your material worked on the C18. I am currently thinking about doing this approach with analtechs hydrocarbon impregnated reverse phase material. I would love to hear your procedures and how it worked for you.
Long time since anyone has submitted anything on this site but thought I Might give it a shot.
Robin I wanted to know how your RP dry column worked out for you. I am curious to use analytechs expensive hydrocarbon impregnated silica in a Dry Vac system as they describe. If you are still linked to the blog can you describe your procedures or have any literature to visit before preforming this purification.
Say, Daniel, why don't you shoot a short film about doing DCVC and post it on youtube? There are a bunch of videos on column chromatography, HPLC and the sort, but not a single one on this technique.
@Beniamin, that is a really good idea. I will try to find the time for it and publish like you suggest. D!
Apropos of the byplay between Robin and Pete, I'm looking to do some DCVC using reverse phase silica (C18). I'm doing a natural products startup, and we're shy the cash to buy a combiflash or really any other major piece of equipment. I've done the HPLC, but I want to move from milligram to gram quantity separations. I've been trying to proof the mobile phases on C18 TLC plates, but the compounds elute between about 15 and 25% acetonitrile on HPLC - and the high water content makes for slow and poor separations by TLC. Sigh.
My questions: any issues with doing DCVC with C18 besides the difficulty packing a good column due to particle size? Have people had pretty good luck transferring a protocol from HPLC to DCVC? If something elutes from HPLC at, say, 15% acetonitrile, is it likely to stay on a DCVC column when you pass 10% over it, and come off when you elute with 15%?
I'm also doing standard underivatized silica gel DCVC, and I've done a series of TLC analyses that define my compounds-of-interest's Rf values in various solvent systems. What kind of Rf am I shooting for? Around 0.5? Less?
Thanks for any responses,
Richard
Hello Y'all;
Not sure I need to resend this, but it didn't post last time.
I'm going to do some DCVC using C18. I'm working on a natural products separation as part of my startup, and lack funds to buy any fancy toys - so it's gonna be old school or nuttin'.
I do know the behavior of my compounds of interest on HPLC, where they elute in the range of 15 - 30% acetonitrile. I've tried to proof the separation by C18 TLC, but high-water mobile phases work like crap with TLC plates. My question: can I use the results of HPLC to accurately predict the behavior on a DCVC column? If I wash with 10% acetonitrile, are the compounds that normally elute at 15% gonna stay, and then come off in 15%? Or is this more empirical and unpredictable?
Any other tips, suggestions, &etc.?
Richard
In Oregon it is legal to use marijuanna extractions but those produced posess irritants and contaminates. I used your method to very quickly, albeit crudely, remove most of what I do not want. In the video the green fraction yields a sticky black tar that smells aweful after solvent purge. The middle fraction demonstrated yielded the bulk of material which is sticky but runs at room temperature and is pale yellow. I am a patient and use this medicine to relieve peripheral nueropathy. I inhale the vapor from a vaporizer to ingest and the pale yellow oil is indeed the extremely potent medicine that I target.
The first fraction yielded much less compound and is bright yellow to orange, but I am puzzled because there was far more material than I anticipated. I believe that I captured some of the middle fraction. I limit myself to three fractions so perhaps I judged when to change flasks incorrectly. I am a patient and a hobbyist only.
https://youtu.be/6A6CXRbKJeA
Anyone familiar with the SARA fraction in fuel oils? Has anyone tried?
Good day, everyone!
I have just discovered this technique and needless to say: I am absolutely blown away! In my clumsy attempts I believe I have managed to succeed in separation. However I have several questions:
1. Is there any reliable way to know how many mls of elution should be collected? How does the diameter of sintered funnel relates to grams of compound to seperate?
2. Should the column be eluted to dryness for each step of gradient elution? I have seen some small thin channels on the bottom of my column. Could the reason be the fact that I have let the wet column stay wet for a couple of minutes?
3. What type of Celite should I use? Does it really matter what particle size of Celite to use?
4. What would be the best way to improve purity? To decrease fraction size? To separate with the same gradient solvent mixture, but with smaller increments? To change the solvent mixture completely? Or to even change the stationary phase to something else?
Any feedback is appreciated!
Sincerely, Arkady.
Hi Arkady,
I'm thrilled to hear about your success with dry column vacuum chromatography! In response to your questions:
1. Elution volumes typically depend on the compound amount being separated. The diameter of the sintered funnel can impact this as well, with larger diameters being suitable for larger amounts of compounds.
2. It's not necessary to elute the column to dryness at each step of gradient elution. The small channels at the bottom could be due to the column staying wet for an extended period. Proper drying is crucial.
3. The type of Celite chosen does impact the filtration quality. The particle size of Celite can also play a role in the effectiveness of the process.
4. To improve purity, consider adjusting the fraction size, altering the gradient solvent mixture incrementally, or changing the stationary phase. Experimentation may be necessary to find the most effective method for your specific separation needs.
Keep up the excellent work, and feel free to reach out with any more questions!
Best,
Irfan Pharmacist
Good Evening, Irfan!
Today I have managed to successfully separate my first compound via DCVC (approx. 90%+ purity by HPLC, I'll need to double check)! Although there were complications due to my clumsiness: I have put too much force into compacting my column... My sintered funnel cracked and then a small shard broke apart...
I believe I have discovered the reason for my new channels formed at the very top of my packed column: too much force led to disruption of structure of silica. I may be wrong, but I can only keep going!
It was a blast nonetheless!
Sincerely, Arkady.
Post a Comment